Research
Cancer is typically considered a genetic disease. However, recent progress in our understanding of epigenetic aberrations in cancer has challenged this view. Overarching goal of our lab is to understand epigenetic mechanisms of transcriptional addiction in cancer and exploit this information to advance cancer therapeutics.
To pursue this objective, we use cutting-edge chromatin conformation capture, high-content Oligopaint DNA FISH, single-cell epigenomics, functional genomics, and combine these technologies with our expertise in computational sciences to systematically explore: i) how epigenetic control of gene expression is disrupted in cancer, ii) why transcriptional addiction can develop, and iii) how heterogeneity and plasticity of transcriptional dependencies enable drug resistance.
1. Understanding driver of genome misfolding in cancer.
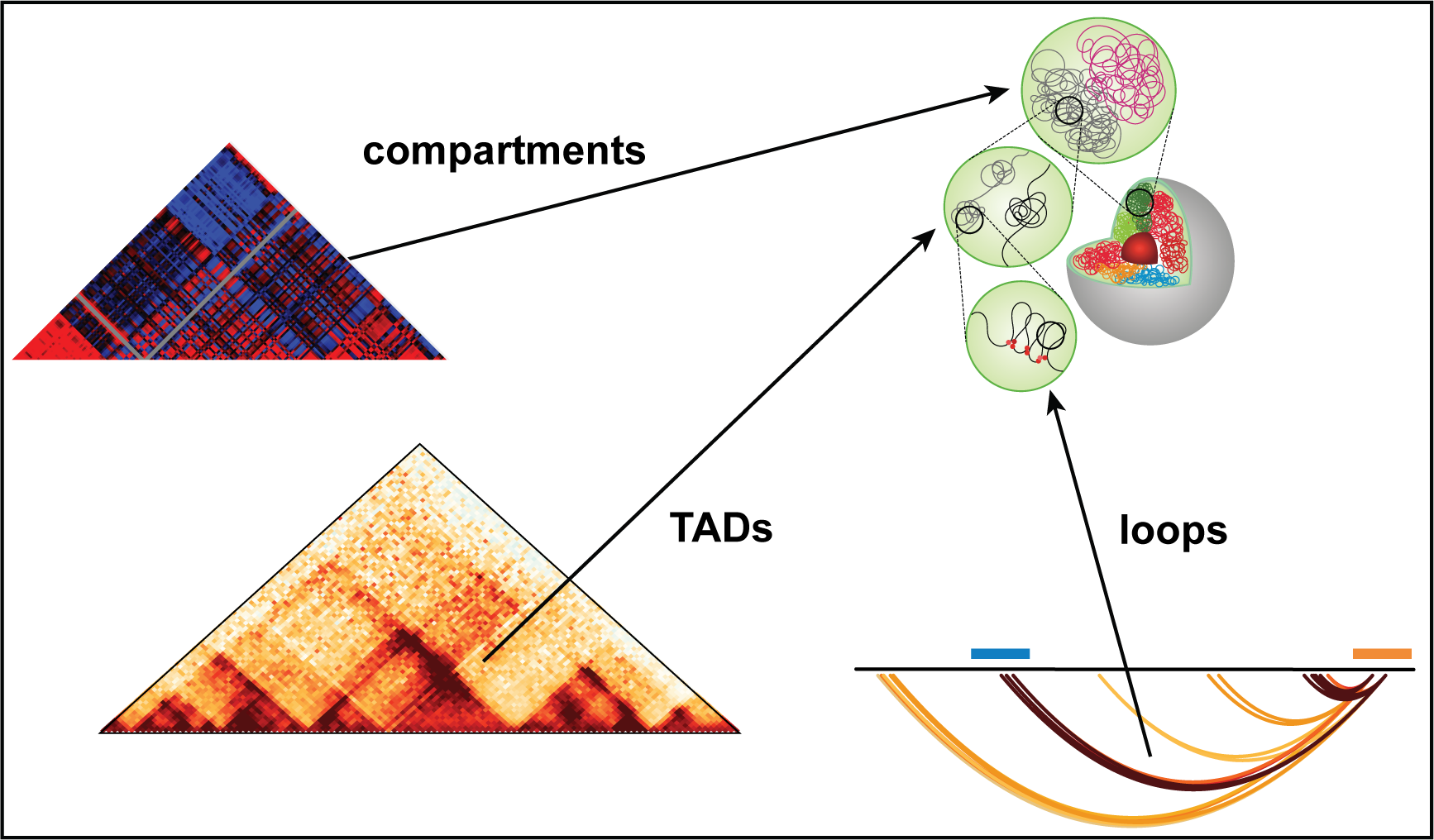
Our lab deploys data-rich experimental techniques in combination with novel computational methods to elucidate epigenetic drivers of transcriptional addiction in Notch-mutated cancers. By measuring Notch impact on chromatin activity and topology, we create the highest possible resolution map of Notch-dependent regulatory maps. Using genetic approaches, we interrogate these maps to elucidate how aberrant epigenetic regulator promotes tumor growth and survival. These studies identify precise epigenetic vulnerabilities of cancer cells and guide treatments of Notch-addicted malignancies. Refer to our manuscripts at Molecular Cell and Cell Reports for more details about our projects.
2. Elucidating the role of epigenetic heterogeneity and plasticity in drug resistance.
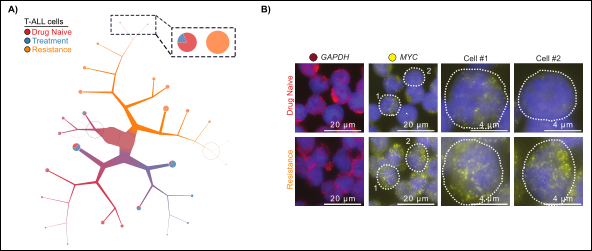
Targeting oncogenic driver of cancers, such as Notch inhibitors, commonly leads to drug resistance. Mechanisms of acquiring resistance to oncology drugs mostly remain unknown, partly due to the limitations of population-based assays in elucidating heterogeneity of drug-naive and complexity of drug-induced tumor evolution. Using single-cell genomics and imaging, we investigate how heterogeneity and plasticity of transcriptional dependencies enables resistance to targeted therapeutics such as Notch inhibitors. For more information about these projects refer to Nature Methods and Pathology News.
3. Computational Genomics.
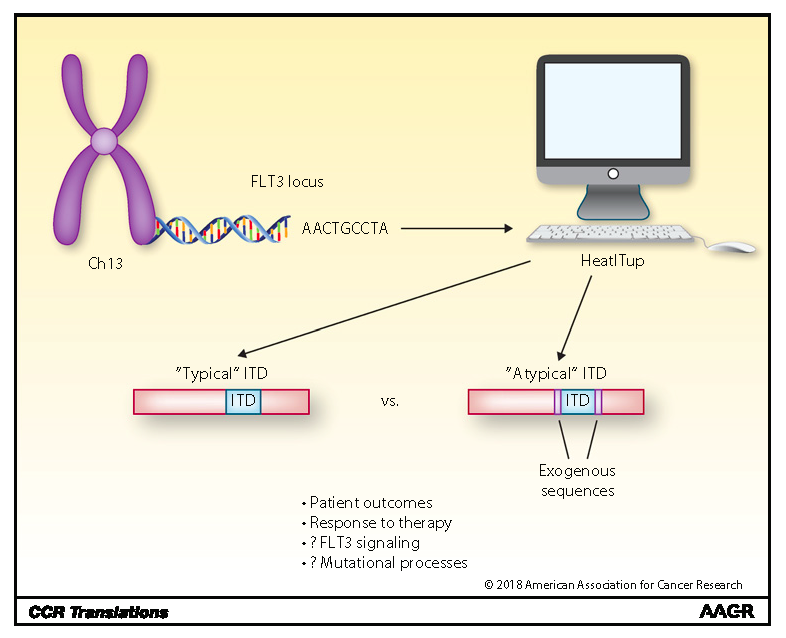
Our lab innovates statistical and machine learning approaches to accelerate discovery of novel therapeutics and biomarkers by elucidating complexity and heterogeneity of tumors. As part of the Center for Personalized Diagnostics, we have access to and mine cancer patient genotypic/phenotypic data sets to improve patient health. To this end, we leverage clinical cases to investigate the correlation between the heterogeneity in mutational structures and response to targeted therapies (see HeatITup algorithm). We also develop computational ecosystems to study molecular and cellular heterogeneity (see TooManyCells algorithm).